Over the last ten years, cancer therapies have struggled with drug resistance. In this report, we explore new coumarin (COU) compounds designed to inhibit the enzyme NQO1, which shows potential for effective treatment due to their favorable predicted drug properties. Three-dimensional (3D) models of NQO1-COUx complexes were generated through in situ modifications of the crystal structure of NQO1-COU12 (PDB entry code: 3JSX), which served as the reference compound for a training set of of 22 and a validation set of 6 VCOUs with known experimental inhibitory potencies. To identify the active conformation of COU1-22, we developed a gas-phase quantitative structure-activity relationship (QSAR) model that established a linear correlation between the calculated enthalpy of NQO1-COU complex formation and the values of experimental activities. Subsequently, we screened the Virtual Compound Library (VCL) using Lipinski's Rule of Five and the PH4 model, then assessed the potency of the new COU analogues using the retained QSAR model. The pharmacokinetic profile of the analogues obtained was also evaluated using the linear correlation equation derived from the QSAR model. The coefficient of determination (R²), the Leave One Out (LOO) cross-validated Squared and the Standard error of regression σ for this equation are 0.91, 0.94 and 0.14, respectively, thus revealing the high predictive power of this model. Similarly, the PH4 model, with a correlation coefficient of 0.91, demonstrated robust predictive power. A comprehensive screening of the COU virtual analogue library yielded a total of 63 drug candidates with oral bioavailability, among which the most promising compounds exhibited a predicted potency of 12.22 and a favorable pharmacokinetic profile. The integration of Quantitative Structure-Activity Relationship (QSAR) techniques and in silico screening, based on the PH4 model, has enabled us to propose potent anticancer candidates with optimal pharmacokinetic profiles.
| Published in | Science Journal of Chemistry (Volume 13, Issue 4) |
| DOI | 10.11648/j.sjc.20251304.12 |
| Page(s) | 102-121 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Coumarins, NQO1, Drug Resistance, Anticancer Therapy, Virtual Screening, Pharmacokinetics
| ||||||
|---|---|---|---|---|---|---|
Compound identification | R5 | R6 | R7 | R8 | X | (nM) |
1 | H | H | H | H | H | 2,6 |
2 | H | CH3 | H | H | H | 14,00 |
3 | OCH3 | H | H | H | H | 2,80 |
4 | H | OCH3 | H | H | H | 11,00 |
5 | H | H | OCH3 | H | H | 6,00 |
6 | H | F | H | H | H | 4,50 |
7 | H | H | F | H | H | 3,80 |
8 | H | Cl | H | H | H | 9,00 |
9 | H | Br | H | Br | H | 4,80 |
10 | H | CH3 | CH3 | H | H | 0,41 |
12 | H | H | C4H4 | C4H4 | H | 0,18 |
13 | H | H | CH3 | CH3 | H | 0,42 |
14 | H | H | H | H | CO2Et | 1221,00 |
15 | H | H | H | H | C3H7 | 588,00 |
| ||||||
|---|---|---|---|---|---|---|
Compound identification | R5 | R6 | R7 | R8 | X | (nM) |
16 | H | CH3 | CH3 | H | 1-naphthyl | 7,70 |
17 | H | CH3 | CH3 | H | 2-naphthyl | 2,50 |
18 | H | CH3 | CH3 | H | phenyl | 39,00 |
19 | H | H | C4H4 | C4H4 | 1-naphthyl | 6,30 |
20 | H | H | C4H4 | C4H4 | 2-naphthyl | 2,20 |
21 | H | H | C4H4 | C4H4 | phenyl | 35,00 |
22 | C4H4 | C4H4 | H | H | 1-naphthyl | 6,30 |
23 | C4H4 | C4H4 | H | H | 2-naphthyl | 6,00 |
24 | H | C4H4 | H | H | phenyl | 15,00 |
25 | H | H | H | H | 1-naphthyl | 24,00 |
26 | H | H | H | H | 2-naphthyl | 14,00 |
27 | H | H | H | H | phenyl | 144,00 |
28 | H | H | H | H | 3,4-diméthyl phenyl | 31,00 |
29 | H | CH3 | CH3 | H | 3,4-diméthyl phenyl | 9,90 |
Training Set | MW [g.mol-1] | ∆∆HMM [kca.mol-1] | ∆∆Gsol [kca.mol-1] | ∆∆TSvib [kca.mol-1] | ∆∆Gcom [kca.mol-1] |
| ||
|---|---|---|---|---|---|---|---|---|
New id. | Old id. [ 4] | |||||||
COU1 | 12 | 0.18 | 436 | 0.00 | 0.00 | 0.00 | 0.00 | 9.74 |
COU2 | 1 | 2.60 | 336 | 3.32 | 1.29 | 0.40 | 4.21 | 8.59 |
COU3 | 2 | 14 | 364 | 8.76 | -2.21 | 1.34 | 5.22 | 7.85 |
COU4 | 3 | 2.80 | 396 | 2.28 | 1.10 | 1.11 | 2.27 | 8.55 |
COU5 | 4 | 11 | 396 | 4.41 | 0.74 | 0.68 | 4.47 | 7.96 |
COU6 | 6 | 4.50 | 372 | 2.48 | -0.59 | -2.15 | 4.03 | 8.35 |
COU7 | 7 | 3.80 | 372 | 6.35 | -3.52 | 0.44 | 2.39 | 8.42 |
COU8 | 10 | 0.41 | 392 | 0.01 | 1.75 | 3.91 | -2.14 | 9.39 |
COU9 | 13 | 0.42 | 392 | 0.15 | -0.44 | 0.26 | -0.55 | 9.38 |
COU10 | 14 | 1221 | 408 | 14.24 | -4.37 | -0.80 | 10.66 | 5.91 |
COU11 | 15 | 588 | 378 | 13.79 | -1.46 | 2.54 | 9.78 | 6.23 |
COU12 | 16 | 7.70 | 318 | 9.48 | -2.02 | 3.93 | 3.53 | 8.11 |
COU13 | 17 | 2.50 | 318 | 6.73 | -1.35 | 3.55 | 1.83 | 8.60 |
COU14 | 18 | 39 | 280 | 10.58 | -1.59 | 2.74 | 6.24 | 7.41 |
COU15 | 20 | 2.20 | 352 | 2.56 | 0.66 | 2.36 | 0.86 | 8.66 |
COU16 | 21 | 35 | 302 | 11.6 | -2.63 | 2.04 | 6.39 | 7.46 |
COU17 | 24 | 15 | 302 | 6.79 | -1.86 | 1.66 | 3.27 | 7.82 |
COU18 | 25 | 24 | 302 | 6.47 | 0.91 | 3.08 | 4.30 | 7.62 |
COU19 | 26 | 14 | 302 | 7.89 | -0.67 | 2.61 | 4.61 | 7.85 |
COU20 | 27 | 144 | 252 | 12.15 | -1.84 | 2.12 | 8.19 | 6.84 |
COU21 | 28 | 31 | 280 | 9.92 | -1.15 | 3.58 | 5.20 | 7.51 |
COU22 | 29 | 9.90 | 308 | 8.44 | -1.02 | 4.51 | 2.91 | 8.00 |
Validation Set | MW [g.mol-1] | ∆∆HMM [kca.mol-1] | ∆∆Gsol [kca.mol-1] | ∆∆TSvib [kca.mol-1] | ∆∆Gcom [kca.mol-1] | / p | ||||
|---|---|---|---|---|---|---|---|---|---|---|
New id. | Old id. [4] | (nM) | ||||||||
VCOU1 | 5 | 6.00 | 8.22 | 396 | 2.47 | 0.68 | 0.32 | 2.83 | 8.34 | 1.01 |
VCOU2 | 8 | 9.00 | 8.05 | 405 | 2.74 | -0.41 | -2.97 | 5.30 | 7.64 | 0.95 |
VCOU3 | 9 | 4.80 | 8.32 | 652 | -2.34 | -3.13 | -8.78 | 3.30 | 8.21 | 0.99 |
VCOU4 | 19 | 6.30 | 8.20 | 352 | 6.86 | -0.56 | 2.67 | 3.63 | 8.11 | 0.99 |
VCOU5 | 22 | 6.30 | 8.20 | 352 | 6.05 | -0.91 | 2.04 | 3.11 | 8.26 | 1.01 |
VCOU6 | 23 | 6.00 | 8.22 | 352 | 7.65 | -2.06 | 1.36 | 4.24 | 7.94 | 0.97 |
Statistical Data of Linear Regression | A | B |
|---|---|---|
= - 0,1981×∆∆HMM + 9,3435 A | ||
= - 0,2867×∆∆Gcom + 9,1546 B | ||
Number of compound n | 22 | 22 |
Squared correlation coefficient of | 0.85 | 0.91 |
LOO cross-validated Squared | 0.86 | 0.94 |
Standard error of regression | 0,23 | 0.14 |
Statistical significance of regression. | 224.1 | 584.3 |
Level of statistical significance | > 95 | > 95 |
Range of activities [nM] | 0.18 –1221 | |
Hypothesis | RSMDa | R2 b | Total Costc | Costs Differenced | Closest Randome | Featuresf |
|---|---|---|---|---|---|---|
Hypo 1 | 2.896 | 0.95 | 145.5 | 893.2 | 192.1 | HBD, HYD-Ar, HYD, HYD, R-Ar |
Hypo 2 | 3.314 | 0.94 | 173.0 | 865.7 | 314.6 | HBD, HYD-Ar, HYD, HYD, R-Ar |
Hypo 3 | 3.416 | 0.93 | 180.5 | 858.2 | 351.2 | HBD, HYD-Ar, HYD, HYD, R-Ar |
Hypo 4 | 3.618 | 0.93 | 196.2 | 842.5 | 388.4 | HBD, HYD-Ar, HYD-Ar, HYD, R-Ar |
Hypo 5 | 3.634 | 0.92 | 197.4 | 841.3 | 425.7 | HBD, HYD, HYD, HYD, R-Ar |
Hypo 6 | 3.937 | 0.91 | 222.7 | 816.0 | 426.5 | HBD, HYD, HYD, HYD, R-Ar |
Hypo 7 | 4.004 | 0.90 | 228.5 | 810.2 | 439.1 | HBD, HYD-Ar, HYD, HYD, R-Ar |
Hypo 8 | 4.069 | 0.91 | 234.2 | 804.5 | 459.5 | HBD, HYD-Ar, HYD, HYD, R-Ar |
Hypo 9 | 4.569 | 0.88 | 282.2 | 756.5 | 479.4 | HBD, HYD-Ar, HYD-Ar, HYD, HYD |
Hypo 10 | 4.659 | 0.87 | 291.7 | 747.0 | 520.3 | HBD, HYD-Ar, HYD-Ar, HYD |
Fixed Cost | 0 | 0 | 52.14 | |||
Null Cost | 9.53 | 0 | 1038.78 |
| |||||
|---|---|---|---|---|---|
New analogues | |||||
C1F | 3-[(7-amino-4-hydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-8-ethyl-4-hydroxy-7-vinyl-chromen-2-one | C2F | 3-[(8-ethyl-4,7-dihydroxy-2-oxo-chromen-3-yl)methyl]-4-hydroxy-7-methyl-8-vinyl-chromen-2-one | C3F | 3-[(8-ethyl-4-hydroxy-7-nitroso-2-oxo-chromen-3-yl)methyl]-4-methoxy-7-methyl-8-vinyl-chromen-2-one |
C4F | 3-[(7-acetyl-4-hydroxy-8-methyl-2-oxo-chromen-3-yl)methyl]-7-ethyl-4-hydroxy-8-methyl-chromen-2-one | C5F | 3-[(8-ethyl-4-hydroxy-2-oxo-7-vinyl-chromen-3-yl)methyl]-4-methoxy-8-methyl-7-vinyl-chromen-2-one | C7F | 8-ethyl-4-hydroxy-3-[[4-methoxy-8-(methylamino)-2-oxo-7-vinyl-chromen-3-yl]methyl]-7-vinyl-chromen-2-one |
C9F | 3-[(8-ethyl-4-hydroxy-7-nitroso-2-oxo-chromen-3-yl)methyl]-4-methoxy-7-methyl-8-vinyl-chromen-2-one | C10F | 3-[(7-amino-8-ethyl-4-hydroxy-2-oxo-chromen-3-yl)methyl]-4-hydroxy-8-phosphanyl-7-propyl-chromen-2-one | C13F | 3-[(8-acetyl-4-hydroxy-2-oxo-7-vinyl-chromen-3-yl)methyl]-4-hydroxy-8-methyl-7-propyl-chromen-2-one |
C14F | 4-hydroxy-3-[(7-hydroxy-4-methoxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-2-oxo-8-propyl-chromene-7-carboxylic acid | C15F | 3-[(4,7-dihydroxy-8-methyl-2-oxo-chromen-3-yl)methyl]-4-hydroxy-2-oxo-7-vinyl-chromene-8-carboxylic acid; ethane | C17F | 3-[(8-ethyl-4-hydroxy-2-oxo-7-vinyl-chromen-3-yl)methyl]-4-methoxy-8-methyl-2-oxo-chromene-7-carbaldehyde |
C18F | 4-hydroxy-3-[(4-hydroxy-7-methyl-2-oxo-8-propyl-chromen-3-yl)methyl]-2-oxo-8-vinyl-chromene-7-carbaldehyde | C19F | 4-hydroxy-3-[(4-hydroxy-8-methyl-2-oxo-7-phosphanyl-chromen-3-yl)methyl]-2-oxo-7-propyl-chromene-8-carboxamide | C20F | 3-[(8-acetyl-4-hydroxy-2-oxo-7-vinyl-chromen-3-yl)methyl]-4-methoxy-8-methyl-7-vinyl-chromen-2-one |
C23F | 7-acetyl-4-hydroxy-3-[(7-hydroxy-4-methoxy-2-oxo-8-phosphanyl-chromen-3-yl)methyl]-8-propyl-chromen-2-one | C25F | 4-hydroxy-3-[(4-hydroxy-8-methyl-7-nitroso-2-oxo-chromen-3-yl)methyl]-7-methyl-8-sulfanyl-chromen-2-one | C26F | 8-ethyl-4-hydroxy-3-[(4-methoxy-7,8-dimethyl-2-oxo-chromen-3-yl)methyl]-2-oxo-chromene-7-carbaldehyde |
C27F | 4-hydroxy-3-[(4-hydroxy-2-oxo-8-phosphanyl-7-propyl-chromen-3-yl)methyl]-7-methyl-8-vinyl-chromen-2-one | C28F | 7-ethyl-4-hydroxy-3-[(4-hydroxy-2-oxo-8-phosphanyl-7-propyl-chromen-3-yl)methyl]-2-oxo-chromene-8-carboxamide | C29F | 8-(1-aminovinyl)-4-hydroxy-3-[(4-hydroxy-2-oxo-7,8-divinyl-chromen-3-yl)methyl]-7-methyl-chromen-2-one; hydrate |
C30F | 3-[(7-ethyl-4-hydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-4-hydroxy-2-oxo-7-phosphanyl-chromene-8-carboxylic acid | C31F | 8-ethyl-3-[(7-hydrosulfinyl-4-hydroxy-8-isopropenyl-2-oxo-chromen-3-yl)methyl]-4-hydroxy-7-sulfanyl-chromen-2-one | C33F | 4,7-dihydroxy-3-[(4-hydroxy-2-oxo-7-phosphanyl-8-propyl-chromen-3-yl)methyl]-8-vinyl-chromen-2-one |
C34F | 3-[(7-amino-8-hydrosulfinyl-4-hydroxy-2-oxo-chromen-3-yl)methyl]-4-hydroxy-7-methyl-8-vinyl-chromen-2-one | C35F | 4,7-dihydroxy-3-[(4-hydroxy-2-oxo-8-propyl-7-sulfanyl-chromen-3-yl)methyl]-8-phosphanyl-chromen-2-one | C36F | 8-acetyl-3-[(8-amino-4-hydroxy-2-oxo-7-vinyl-chromen-3-yl)methyl]-7-ethyl-4-hydroxy-chromen-2-one |
C37F | 7-ethyl-3-[(7-formyl-4-hydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-4-hydroxy-2-oxo-chromene-8-carboxylic acid | C38F | 4-hydroxy-3-[(4-hydroxy-2-oxo-8-phosphanyl-7-propyl-chromen-3-yl)methyl]-2-oxo-7-sulfanyl-chromene-8-carboxylic acid | C39F | ammonia; 3-[(8-ethyl-4-hydroxy-2-oxo-7-vinyl-chromen-3-yl)methyl]-4-methoxy-7-methyl-8-vinyl-chromen-2-one |
C40F | 4,7-dihydroxy-3-[(4-hydroxy-8-nitroso-2-oxo-7-sulfanyl-chromen-3-yl)methyl]-8-vinyl-chromen-2-one | C41F | 8-ethyl-4-hydroxy-3-[(7-hydroxy-4-methoxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-7-nitroso-chromen-2-one | C42F | 3-[(7-amino-4-hydroxy-2-oxo-8-phosphanyl-chromen-3-yl)methyl]-4-hydroxy-2-oxo-7-vinyl-chromene-8-carbaldehyde |
C43F | 3-[(7-acetyl-4-hydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-4-hydroxy-8-nitroso-7-propyl-chromen-2-one | C45F | 8-formyl-3-[(4-hydroxy-8-methyl-2-oxo-7-vinyl-chromen-3-yl)methyl]-4-methoxy-2-oxo-chromene-7-carboxamide | C46F | 3-[(4,7-dihydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-8-hydrosulfinyl-4-hydroxy-7-propyl-chromen-2-one |
C51F | 3-[(8-ethyl-4-hydroxy-7-nitroso-2-oxo-chromen-3-yl)methyl]-4-methoxy-7-methyl-8-vinyl-chromen-2-one | C52F | 4-hydroxy-3-[(4-hydroxy-2-oxo-8-phosphanyl-7-sulfanyl-chromen-3-yl)methyl]-2-oxo-8-vinyl-chromene-7-carbaldehyde | C53F | 3-[(7-ethyl-4-hydroxy-2-oxo-8-sulfanyl-chromen-3-yl)methyl]-4-hydroxy-2-oxo-8-vinyl-chromene-7-carboxylic |
C54F | 7-ethyl-3-[(7-formyl-4-hydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-4-hydroxy-2-oxo-chromene-8-carboxylic | C55F | 3-[(7-amino-4-hydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-4-hydroxy-7-methyl-8-vinyl-chromen-2-one | C56F | 4-hydroxy-3-[(4-hydroxy-2-oxo-8-sulfanyl-7-vinyl-chromen-3-yl)methyl]-2-oxo-chromene-7,8-dicarbaldehyde |
C57F | 3-[(4,7-dihydroxy-8-methyl-2-oxo-chromen-3-yl)methyl]-4,7-dihydroxy-2-oxo-chromene-8-carbaldehyde | C587 | 3-[(4,8-dihydroxy-2-oxo-7-vinyl-chromen-3-yl)methyl]-4-hydroxy-8-phosphanyl-7-propyl-chromen-2-one | C60F | 3-[(4,8-dihydroxy-2-oxo-7-phosphanyl-chromen-3-yl)methyl]-4-hydroxy-8-phosphanyl-7-propyl-chromen-2-one |
C61F | 3-[(8-formyl-4-hydroxy-2-oxo-7-phosphanyl-chromen-3-yl)methyl]-4-hydroxy-2-oxo-8-phosphanyl-chromene-7-carbaldehyde | C63F | 3-[(4,8-dihydroxy-7-methyl-2-oxo-chromen-3-yl)methyl]-4-hydroxy-7-methyl-8-vinyl-chromen-2-one | C64F | 3-[(4,8-dihydroxy-2-oxo-7-phosphanyl-chromen-3-yl)methyl]-4-hydroxy-8-nitroso-7-phosphanyl-chromen-2-one |
C73F | 3-[(7-formyl-4-hydroxy-2-oxo-8-sulfanyl-chromen-3-yl)methyl]-4,8-dihydroxy-2-oxo-chromene-7-carbaldehyde | C76F | 4-hydroxy-3-[(4-hydroxy-2-oxo-8-phosphanyl-7-propyl-chromen-3-yl)methyl]-7-methyl-8-vinyl-chromen-2-one | C78F | 7-ethyl-4-hydroxy-3-[(4-hydroxy-2-oxo-8-phosphanyl-7-propyl-chromen-3-yl)methyl]-2-oxo-chromene-8-carboxamide |
C81F | 3-[(8-ethyl-4,7-dihydroxy-2-oxo-chromen-3-yl)methyl]-4-hydroxy-8-nitroso-7-vinyl-chromen-2-one | C82F | 7-ethyl-4-hydroxy-3-[(4-hydroxy-8-nitroso-2-oxo-7-phosphanyl-chromen-3-yl)methyl]-8-vinyl-chromen-2-one | C83F | 4-hydroxy-3-[(4-hydroxy-8-methyl-2-oxo-7-propyl-chromen-3-yl)methyl]-2-oxo-8-phosphanyl-chromene-7-carboxylic |
C85F | 3-[(4,7-dihydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-7-ethyl-4-hydroxy-8-nitroso-chromen-2-one | C89F | 3-[(4,7-dihydroxy-8-nitroso-2-oxo-chromen-3-yl)methyl]-4-hydroxy-8-phosphanyl-7-propyl-chromen-2-one | C91F | 4-hydroxy-3-[(4-hydroxy-2-oxo-7-propyl-8-sulfanyl-chromen-3-yl)methyl]-2-oxo-8-phosphanyl-chromene-7-carboxylic |
C92F | 3-[(8-acetyl-4-hydroxy-7-nitroso-2-oxo-chromen-3-yl)methyl]-4-methoxy-7-methyl-8-phosphanyl-chromen-2-one | C93F | 4-hydroxy-3-[(4-hydroxy-2-oxo-8-phosphanyl-7-propyl-chromen-3-yl)methyl]-2-oxo-7-sulfanyl-chromene-8-carboxylic | C96F | 3-[(4,7-dihydroxy-2-oxo-8-vinyl-chromen-3-yl)methyl]-4-hydroxy-7-propyl-8-sulfanyl-chromen-2-one |
C98F | 7-ethyl-4-hydroxy-3-[(4-hydroxy-2-oxo-7-phosphanyl-8-sulfanyl-chromen-3-yl)methyl]-8-vinyl-chromen-2-one | C100 | 8-acetyl-4-hydroxy-3-[(4-hydroxy-2-oxo-7-propyl-8-sulfanyl-chromen-3-yl)methyl]-7-methyl-chromen-2-one | C108F | 3-[(7-amino-8-hydrosulfinyl-4-hydroxy-2-oxo-chromen-3-yl)methyl]-4-hydroxy-7-propyl-8-sulfanyl-chromen-2-one |
N | Analogs | MW [g.mol-1] | ∆∆HMM [kca.mol-1] | ∆∆Gsol [kca.mol-1] | ∆∆TSvib [kca.mol-1] | ∆∆Gcom [kca.mol-1] | |
|---|---|---|---|---|---|---|---|
C1 | F1-F8-F8-F6 | 431 | 3.55 | -0.75 | 5.14 | -2.35 | 9.83 |
C2 | F1- F10- F8- F2 | 420 | -0.08 | 1.97 | 2.41 | -0.53 | 9.31 |
C3 | F1-F13-F8-F10 | 435 | 4.79 | -0,43 | -0.37 | 4.73 | 7.80 |
C4 | F2-F1-F2-F4 | 434 | -1,36 | 2.51 | 5.85 | -4.70 | 10.50 |
C5 | F1-F8-F2-F9 | 432 | 4.18 | -0.75 | 0.43 | 2.99 | 8.30 |
C7 | F1-F8-F6-F8 | 458 | 3.23 | -0.55 | 4.54 | -1.85 | 9.69 |
C9 | F1-F13-F8-F6 | 434 | 2.69 | -0.47 | 2.12 | 0.11 | 9.12 |
C10 | F1-F6-F14-F3 | 453 | -3.41 | 3.17 | 8.74 | -8.99 | 11.73 |
C13 | F2-F3-F4-F8 | 460 | -2.64 | 3.53 | 3.59 | -2.70 | 9.93 |
C14 | F3-F7-F8-F10 | 464 | 3.06 | 1.62 | 1.31 | 3.37 | 8.19 |
C15 | F2-F10-F7-F8 | 436 | -0.27 | 2.13 | -1.29 | 3.16 | 8.25 |
C17 | F2-F9-F1-F8 | 432 | 4.17 | -0.74 | 0.40 | 3.04 | 8.28 |
C18 | F3-F2-F8-F9 | 446 | 3.51 | 2,19 | 3.46 | 2.24 | 8.51 |
C19 | F2-F14-F5-F3 | 467 | -2.68 | 3.38 | 0.09 | 0.60 | 8.98 |
C20 | F2-F8-F4-F8 | 444 | 2.11 | -0.68 | 2.82 | -1.39 | 9.55 |
C23 | F3-F4-F14-F10 | 468 | 3.10 | 0.49 | 5.27 | -1.68 | 9.64 |
C25 | F2-F13-F16-F2 | 425 | 0.66 | 2.25 | -1.33 | 4.24 | 7.94 |
C26 | F2-F6-F1-F9 | 421 | 2.71 | -0.19 | 0.38 | 2.14 | 8.54 |
C27 | F8-F2-F14-F3 | 450 | -2.84 | 2.99 | 2.06 | -1.91 | 9.70 |
C28 | F5-F1-F14-F3 | 481 | -1.90 | 1.93 | 3.91 | -3.87 | 10.26 |
C29 | F5-F2-F8-F8 | 445 | -0.96 | 3.26 | 3.19 | -0.89 | 9.41 |
C30 | F8-F1-F7-F14 | 466 | -2.33 | 2.71 | -17.93 | 18.32 | 3.90 |
C31 | F4-F15-F1-F16 | 486 | 0.81 | 2.52 | -0.36 | 3.69 | 8.10 |
C33 | F3-F14-F8-F10 | 452 | 4.48 | 2.52 | 4.58 | 2.41 | 8.46 |
C34 | F8-F2-F15-F6 | 439 | 1.81 | 1.64 | 1.55 | 1.89 | 8.61 |
C35 | F3-F16-F14-F10 | 458 | 4.02 | 2.77 | 2.55 | 4.24 | 7.94 |
C36 | F4-F1-F6-F8 | 447 | -3.22 | 2.72 | 2.92 | -3.43 | 10.14 |
C37 | F7-F1-F8-F9 | 462 | -2.04 | 2.19 | -0.61 | 0.76 | 8.94 |
C38 | F7-F16-F14-F3 | 486 | 3.59 | 3.14 | 1.47 | -1.92 | 9.70 |
C39 | F8-F6-F1-F8 | 431 | -5.25 | -0.37 | 5.06 | -10.68 | 12.22 |
C40 | F8-F10-F13-F16 | 439 | 0.75 | 3.00 | 1.07 | 2.68 | 8.39 |
C41 | F8-F10-F1-F13 | 435 | 4.79 | -0.42 | -0.35 | 4.72 | 7.80 |
C42 | F9-F8-F14-F6 | 437 | -0.53 | 2.29 | -0.58 | 2.34 | 8.48 |
C43 | F8-F4-F13-F3 | 475 | -2.49 | 2,68 | -0.16 | 0.34 | 9.06 |
C45 | F9-F5-F2-F8 | 447 | 3.32 | -0.70 | -1.13 | 3.75 | 8.08 |
C46 | F8-F10-F15-F3 | 468 | 0.55 | 3.69 | 1.95 | 2.28 | 8.50 |
C51 | F8-F6-F1-F13 | 434 | 2.69 | 1.45 | 2.12 | 2.03 | 8,57 |
C52 | F8-F9-F14-F16 | 454 | 1.02 | 1.98 | -1.12 | 4,12 | 7.97 |
C53 | F8-F7-F16-F1 | 466 | -1.55 | 2.67 | 0.26 | 0.86 | 8.91 |
C54 | F8-F9-F7-F1 | 462 | -2.02 | 2.21 | -1.21 | 1.40 | 8.75 |
C55 | F8-F6-F8-F2 | 417 | -0.35 | 2.65 | 1.46 | 0.84 | 8.91 |
C56 | F9-F9-F16-F8 | 450 | -0.03 | 2.42 | -0.76 | 3.16 | 8.25 |
C57 | F9-F10-F2-F10 | 410 | 3.25 | 2.14 | -1.35 | 6.74 | 7.22 |
C58 | F10-F8-F14-F3 | 452 | -3.50 | 2.99 | 4.86 | -5,37 | 10.69 |
C60 | F10-F14-F14-F3 | 458 | -3.54 | 3.19 | 5.10 | -5.45 | 10.72 |
C61 | F9-F14-F14-F9 | 456 | -1.07 | 2,20 | -1.61 | 2.74 | 8.37 |
C63 | F10-F2-F8-F2 | 406 | 0.25 | 2.52 | 2.30 | 0.47 | 9.02 |
C64 | F10-F14-F13-F14 | 445 | -1.86 | 3.03 | -0.26 | 1,43 | 8.74 |
C73 | F10-F9-F16-F9 | 440 | -0.49 | 4.20 | -1.74 | 5.46 | 7.59 |
C76 | F14-F3-F8-F2 | 450 | -3.41 | 2.86 | 2.41 | -2.95 | 10.00 |
C78 | F14-F3-F5-F1 | 481 | -5.56 | 4.72 | 2.80 | -3.64 | 10.20 |
C81 | F13-F8-F1-F10 | 435 | -0.14 | 2.13 | 1.83 | 0.16 | 9.11 |
C82 | F13-F14-F8-F1 | 451 | -2.46 | 3.27 | 2.00 | -1,18 | 9.49 |
C83 | F14-F7-F2-F3 | 468 | -3.17 | 2.78 | 1.53 | -1.91 | 9.70 |
C85 | F13-F1-F8-F10 | 435 | 1.70 | 2.50 | 0,77 | 3.43 | 8.17 |
C89 | F13-F10-F14-F3 | 455 | -0.90 | 2,94 | 3.79 | -1.74 | 9.65 |
C91 | F14-F7-F16-F3 | 486 | -2.64 | 2.74 | 0.06 | 0.03 | 9.15 |
C92 | F14-F16-F4-F13 | 471 | 1.48 | 0.00 | 0.16 | 1.32 | 8.77 |
C93 | F14-F3-F7-F16 | 486 | -3.41 | 3.12 | 1.43 | -1.71 | 9.65 |
C96 | F16-F3-F8-F10 | 452 | -1,15 | 2.77 | 1.99 | -0.36 | 9.26 |
C98 | F16-F14-F8-F1 | 454 | -0.67 | 2.44 | 2.55 | -0.78 | 9.38 |
C100 | F16-F3-F4-F2 | 466 | -1.67 | 3.14 | 3.77 | -2.31 | 9.82 |
C108 | F15-F6-F16-F3 | 473 | -0.48 | 2.42 | 1.42 | 0.52 | 9.01 |
Ref | COU1 | 436 | 0 | 0 | 0 | 0 | g |
Analoguesa | #starsb | Mwc | Smold | Smolhfoe | Vmolf | #rotBg | HBdonh |
|---|---|---|---|---|---|---|---|
F1-F8-F8-F6 | 0 | 431 | 725.8 | 250.2 | 1299.1 | 8 | 3.5 |
F2-F1-F2-F4 | 0 | 434 | 726.5 | 358.7 | 1306.2 | 6 | 2 |
F1-F6-F14-F3 | 1 | 454 | 747.5 | 293.5 | 1345.1 | 9 | 5.5 |
F2-F3-F4-F8 | 0 | 460 | 768.5 | 367.3 | 1390.2 | 8 | 2 |
F5-F1-F14-F3 | 1 | 481 | 751.3 | 290.4 | 1378.3 | 9 | 6 |
F4-F1-F6-F8 | 0 | 447 | 702.3 | 273.6 | 1293.5 | 8 | 3.5 |
F8-F6-F1-F8 | 0 | 431 | 718.7 | 250.1 | 12978 | 8 | 3.5 |
F10-F8-F14-F3 | 1 | 452 | 730 | 241.1 | 1308 | 9 | 5 |
F10-F14-F14-F3 | 2 | 458 | 724 | 177 | 1289.2 | 9 | 7 |
F14-F3-F8-F2 | 1 | 450 | 714.4 | 301.6 | 1311.2 | 8 | 4 |
F14-F3-F5-F1 | 1 | 481 | 752.2 | 291.1 | 1379.3 | 9 | 6 |
F16-F3-F4-F2 | 0 | 466 | 734.5 | 322.6 | 1331.1 | 8 | 2.8 |
COU1 | 0 | 436 | 666.6 | 167.9 | 1201 | 7 | 4 |
Finastéride | 0 | 372.550 | 605.996 | 431.904 | 1152.042 | 3 | 2.000 |
Alfuzosine | 1 | 389.453 | 919.272 | 417.508 | 1140.426 | 3 | 3.000 |
Téraz (R) | 0 | 387.438 | 621.508 | 422.153 | 1137.212 | 9 | 2.000 |
Téraz (S) | 0 | 387.438 | 603.495 | 405.495 | 1131.925 | 5 | 2.000 |
Analoguesa | HBacci | LogPo/wj | LogSwatk | LogKhsal | LogB/Bm | BIPcacon | #metabo | PIC50prep | HOAq | %HOAr |
|---|---|---|---|---|---|---|---|---|---|---|
F1-F8-F8-F6 | 7.5 | 2.845 | -6.032 | 0.235 | -2.063 | 119.2 | 5 | 9.83 | 3 | 80.8 |
F2-F1-F2-F4 | 8.5 | 2.651 | -5.822 | 0.230 | -1.870 | 135.2 | 6 | 10.50 | 3 | 80.6 |
F1-F6-F14-F3 | 7.5 | 2.878 | -6.241 | 0.138 | -1.993 | 117.2 | 7 | 11.73 | 2 | 80.8 |
F2-F3-F4-F8 | 8.5 | 3.290 | -6.337 | 0.349 | -1.928 | 173.6 | 5 | 9.93 | 3 | 86.3 |
F5-F1-F14-F3 | 9 | 2.330 | -6.208 | -0.007 | -2.137 | 79.6 | 7 | 10.26 | 2 | 74.6 |
F4-F1-F6-F8 | 9.5 | 2.047 | -5.621 | -0.057 | -1.936 | 119.9 | 6 | 10.14 | 3 | 76.1 |
F8-F6-F1-F8 | 7.5 | 2.841 | -6.032 | 0.239 | -2.018 | 120.8 | 5 | 12.22 | 3 | 80.8 |
F10-F8-F14-F3 | 7.3 | 3.015 | -6.365 | 0.133 | -1.812 | 158.5 | 6 | 10.69 | 3 | 84 |
F10-F14-F14-F3 | 7.3 | 2.639 | -6.227 | -0.106 | -1.779 | 117 | 6 | 10.72 | 3 | 79.4 |
F14-F3-F8-F2 | 6.5 | 3.830 | -6.706 | 0.396 | -1.162 | 472.2 | 6 | 10.00 | 3 | 100 |
F14-F3-F5-F1 | 9 | 2.344 | -6.208 | -0.005 | -2.129 | 81.2 | 7 | 10.20 | 2 | 74.9 |
F16-F3-F4-F2 | 9 | 2.827 | -6.219 | 0.107 | -1.687 | 190.1 | 6 | 9.82 | 3 | 84.3 |
COU1 | 9.3 | 1.641 | -5.464 | -0.364 | -2.453 | 8.3 | 5 | 2 | 53.0 | |
Finastéride | 5.000 | 3.553 | -4.91 | 0.549 | -0.75 | 777.322 | 1 | 3 | 100 | |
Alfuzosine | 8.700 | 1.322 | -3.639 | -0.601 | -1.076 | 347.334 | 5 | 3 | 80.158 | |
Téraz (R) | 9.200 | 1.273 | -3.425 | -0.452 | -0.646 | 470.273 | 4 | 3 | 82.229 | |
Téraz (S) | 9.200 | 1.262 | -3.425 | -0.440 | -0.596 | 467.119 | 4 | 3 | 82.113 |
2D | Two-dimensional |
3D | Three-dimensional |
ADME | Absorption, Distribution, Metabolism, and Excretion |
COU | Coumarin Inhibitors |
COUx | Coumarin Inhibitors of the Training Set |
VCOUs | Coumarin Inhibitors of the Validation Set |
∆∆Gcom | Relative Complexation Gibbs Free Energy |
GFE | Gibbs Free Energy |
∆∆Gsol | Relative Solvation Gibbs Free Energy |
HBA | Hydrogen Bond Acceptor |
HBD | Hydrogen Bond Donor |
HMM | Enthalpy Component of Gibbs Free Energy |
HOA | Human Oral Absorption |
HYD | Hydrophobic |
HYDA | Hydrophobic Aliphatic |
Ki | Inhibitory Concentration |
MM | Molecular Mechanics |
MM-PB | Molecular Mechanics–poisson–boltzmann |
NQO1 | NAD(P)H Quinone Oxydoréductase 1 |
PDB | Protein Data Bank |
PH4 | Pharmacophore |
QSAR | Quantitative Structure–activity Relationships |
RMSD | Root-mean Square Deviation |
SAR | Structure–activity Relationships |
TS | Training Set |
VS | Validation Set |
| [1] | Xie Y H, Chen Y X, Fang J Y. Comprehensive review of targeted therapy for colorectal cancer. Signal transduction and targeted therapy, 2020, 5(1), 22. |
| [2] | Kolotyeva N A, Groshkov A A, Rozanova N A, Berdnikov A K, Novikova, S V, Komleva, Y K, Piradov, M A. Pathobiochemistry of Aging and Neurodegeneration: Deregulation of NAD+ Metabolism in Brain Cells. Biomolecules, 2024, 14(12), 1556. |
| [3] | Nolan K A, Doncaster J R, Dunstan M S, Scott K A, Frenkel A D, Siegel D, Bryce R A. Synthesis and biological evaluation of coumarin-based inhibitors of NAD (P) H: quinone oxidoreductase-1 (NQO1). Journal of medicinal chemistry, 2009, 52(22), 7142-7156. |
| [4] | Verma S, Pathak R K. Discovery and optimization of lead molecules in drug designing. In Bioinformatics. Academic Press, 2022, 253-267. |
| [5] | Niazi S K, and Mariam Z. Computer-aided drug design and drug discovery: a prospective analysis. Pharmaceuticals, 2023, 17(1), 22. |
| [6] | Araújo R S, Silva-Junior E F, Aquino T M, Scotti M T, Ishiki H M, Scotti L, Mendonça-Junior F J B. Computer-aided drug design applied to secondary metabolites as anticancer agents. Current Topics in Medicinal Chemistry, 2020, 20(19), 1677-1703. |
| [7] | Lionta E, Spyrou G, Vassilatis D K, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Current Topics in Medicinal Chemistry, (2014), 14(16): 1923–1938. |
| [8] | Zhang T, Li X, Kumar R, Chen Y. Future perspective on computer-aided drug design for oncology therapeutics. Pharmacological Innovations, 2024, 32(3): 55-69. |
| [9] | Li R, Bianchet M A, Talalay P, Amzel L M. The three-dimensional structure of NAD (P) H: quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: mechanism of the two-electron reduction. Proceedings of the National academy of sciences, (1995), 92(19): 8846-8850. |
| [10] | Yang C, Hu Q. Roles of NAD(P)H: quinone oxidoreductase 1 (NQO1) in diverse diseases. Life, 2021, 11(12): 1301. |
| [11] | Silva V L, Silva-Reis R, Moreira-Pais A, Ferreira T, Oliveira P A, Ferreira R, Grozea I. Dicoumarol: From chemistry to antitumor benefits. Chinese Medicine, 2022, 17(1), 145. |
| [12] | Kang S Y, Lee J H, Park E J. IP‑DNQ induces mitochondrial dysfunction and ROS‑mediated apoptosis in NQO1‑overexpressing pancreatic cancer cells. Antioxidants & Redox Signaling, 2023. |
| [13] | Berridge M V, Herst P M, Prata C. Cellular reductive stress: Is plasma membrane electron transport an evolutionarily-conserved safety valve? Redox Biochem. Chem., 2023, 5(6): 1-9. |
| [14] | Insight-II and Discover Molecular Modeling and Simulation Package, version 2005; Accelrys: San Diego, CA, USA, 2005. |
| [15] | Discovery Studio molecular modeling and simulation program, Version 2.5, Accelrys, Inc., San Diego, CA, 2009. |
| [16] | Wang X, Xu Y, Zheng H, Yu K. An Extendible, Graph-Neural-Network-Based Approach for Accurate Force Field Development of Large Flexible Organic Molecules. arXiv preprint arXiv: 2106.00927, 2021. |
| [17] | Niaré A., Alex, A. Y. Z., Bernard, D. A. M., Marius, K. S., Stéphane, D. S., Moise, K. A., Guy-Richard, K. M., Fagnidi, Y. K. H., & Doh, S. (2025). Study by Molecular Docking of the Interactions Between Dihydroorotate Dehydrogenase and a Series of Inhibitors of Pyrrole Derivatives for the Treatment of Malaria. Asian Journal of Chemical Sciences, 15(1): 92-110. |
| [18] | Soro I, N’Guessan H, Abou A, N’Guessan R K, Megnassan E. Conformational study of molecules in a biological environment, design of inhibitors of human aminopeptidase m1 implicated in cancer therapy. Universal Journal of Pharmaceutical Research. in our previous studies on the design of inhibitors. Universal J. of Pharm Research, 2023, 8(5): 71-86. |
| [19] | Gilson, Michael K. and Barry H. The inclusion of electrostatic hydration energies in molecular mechanics calculations, J. Comput. Aided Mol. Des., 1991, 5(1), 5-20. |
| [20] | Owono Owono L C, Ntie‐Kang F, Keita M, Megnassan E, Frecer V, Miertus S. Virtually Designed Triclosan‐Based Inhibitors of Enoyl‐Acyl Carrier Protein Reductase of Mycobacterium tuberculosis and of Plasmodium falciparum. Molecular Informatics, 2015, 34(5), 292-307. |
| [21] | QikProp; Version 3.7, Release 14, X Schrödinger; LLC: New York, NY, USA, 2014. |
| [22] | Herbert J M. Dielectric continuum methods for quantum chemistry. WIREs Computational Molecular Science, 2022, 12(1): 1519. |
| [23] | Barber C, Heghes C, Johnston L. A framework to support the application of the OECD guidance documents on (Q) SAR model validation and prediction assessment for regulatory decisions. Computational Toxicology, 2024, 30: 100305. |
| [24] | N’Guessan H, Soro I, Keita M, Megnassan E. Design and In silico Screening of Combinatorial Library of New Herbicidal Analogs of Cycloalka [d] quinazoline 2, 4 dione− Benzoxazinones Inhibiting Protoporphyrinogen IX Oxidase. J Pharm Res Int, 2022, 34(56): 42-61. |
| [25] | Niaré, A., N’guessan, A.-B., Djako, B. A. M., Dembélé, G. S., Koné, M. G.-R., Yéo, Y. (2024). QSAR, pharmacophore, and molecular docking studies for the design of novel arylamide-derived inhibitors of Mycobacterium tuberculosis. Chemical Science International Journal, 33(6), 212–224. |
| [26] | Keita M, Yaya Y, Yvon B M B, Esmel A E, Dali B, N’Guessan, H. Molecular and Thermodynamic Modeling of the Protein-Ligand Interaction. Application to Computer-Assisted Design of Anti-Competitive Inhibi-tors of Human Histone Deacetylase, 2021, 2: 606-630. |
| [27] | Kone M, N’Guessan H, N'gouan A J, Megnassan E. Computer-aided design of new hydroxamic acid derivatives targeting the Plasmodium falciparum M17 Metallo-aminopeptidase with Favorable Pharmacokinetic Profile. Int. J. Pharm. Sci. Drug Res., 2023: 356-375. |
| [28] | Niaré A, N’Guessan H, Dali B, Megnassan E. Computer-assisted design of hydroxamic acid derivatives inhibitors of M1 Metallo Aminopeptidase of Plasmodium falciparum with favorable pharmacokinetic profile. J Pharm Res Int, 2022: 21-44. |
| [29] | Bléhoué I C, Koné M, Esmel E A, Fofana I, Kéïta M, Megnassan E. Molecular modelling and virtual screening application to the computer-aided design of anticancer inhibitors with a favorable pharmacokinetic profile against e6 papillomavirus type 16. Universal Journal of Pharmaceutical Research. design of inhibitors. 2024. |
| [30] | Revillo Imbernon J, Jacquemard C, Bret G, Marcou G, Kellenberger E. Comprehensive analysis of commercial fragment libraries. RSC Medicinal Chemistry, 2022, 13(3): 300–310. |
| [31] | Bernard Djako A M, Adama N, Issouf F, Mathias M L, Mélalie K et Eugène M. Computer-assisted Design of Novel Polyketide Synthase 13 of Mycobacterium tuberculosis Inhibitors Using Molecular Modeling and Virtual Screening. Journal of Advances in Medical and Pharmaceutical Sciences, 2025, 27(6): 1-18. |
| [32] | Davis J M, Smith L R. Advances in computational approaches for estimating passive permeability in drug discovery. Membranes, 2023, 13(11) 851. |
APA Style
Yao, H. K., Abou, A., Djandé, A., Adama, N., Eugène, M., et al. (2025). Computer-aided Design of Coumarin Inhibitors of Quinone Oxidoreductase-1 (NQO1) with a Favorable Pharmacokinetic Profile. Science Journal of Chemistry, 13(4), 102-121. https://doi.org/10.11648/j.sjc.20251304.12
ACS Style
Yao, H. K.; Abou, A.; Djandé, A.; Adama, N.; Eugène, M., et al. Computer-aided Design of Coumarin Inhibitors of Quinone Oxidoreductase-1 (NQO1) with a Favorable Pharmacokinetic Profile. Sci. J. Chem. 2025, 13(4), 102-121. doi: 10.11648/j.sjc.20251304.12
@article{10.11648/j.sjc.20251304.12,
author = {Honoré Kouadio Yao and Akoun Abou and Abdoulaye Djandé and Niaré Adama and Megnassan Eugène and Issouf Soro},
title = {Computer-aided Design of Coumarin Inhibitors of Quinone Oxidoreductase-1 (NQO1) with a Favorable Pharmacokinetic Profile
},
journal = {Science Journal of Chemistry},
volume = {13},
number = {4},
pages = {102-121},
doi = {10.11648/j.sjc.20251304.12},
url = {https://doi.org/10.11648/j.sjc.20251304.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.sjc.20251304.12},
abstract = {Over the last ten years, cancer therapies have struggled with drug resistance. In this report, we explore new coumarin (COU) compounds designed to inhibit the enzyme NQO1, which shows potential for effective treatment due to their favorable predicted drug properties. Three-dimensional (3D) models of NQO1-COUx complexes were generated through in situ modifications of the crystal structure of NQO1-COU12 (PDB entry code: 3JSX), which served as the reference compound for a training set of of 22 and a validation set of 6 VCOUs with known experimental inhibitory potencies. To identify the active conformation of COU1-22, we developed a gas-phase quantitative structure-activity relationship (QSAR) model that established a linear correlation between the calculated enthalpy of NQO1-COU complex formation and the values of experimental activities. Subsequently, we screened the Virtual Compound Library (VCL) using Lipinski's Rule of Five and the PH4 model, then assessed the potency of the new COU analogues using the retained QSAR model. The pharmacokinetic profile of the analogues obtained was also evaluated using the linear correlation equation derived from the QSAR model. The coefficient of determination (R²), the Leave One Out (LOO) cross-validated Squared and the Standard error of regression σ for this equation are 0.91, 0.94 and 0.14, respectively, thus revealing the high predictive power of this model. Similarly, the PH4 model, with a correlation coefficient of 0.91, demonstrated robust predictive power. A comprehensive screening of the COU virtual analogue library yielded a total of 63 drug candidates with oral bioavailability, among which the most promising compounds exhibited a predicted potency of 12.22 and a favorable pharmacokinetic profile. The integration of Quantitative Structure-Activity Relationship (QSAR) techniques and in silico screening, based on the PH4 model, has enabled us to propose potent anticancer candidates with optimal pharmacokinetic profiles.},
year = {2025}
}
TY - JOUR T1 - Computer-aided Design of Coumarin Inhibitors of Quinone Oxidoreductase-1 (NQO1) with a Favorable Pharmacokinetic Profile AU - Honoré Kouadio Yao AU - Akoun Abou AU - Abdoulaye Djandé AU - Niaré Adama AU - Megnassan Eugène AU - Issouf Soro Y1 - 2025/08/04 PY - 2025 N1 - https://doi.org/10.11648/j.sjc.20251304.12 DO - 10.11648/j.sjc.20251304.12 T2 - Science Journal of Chemistry JF - Science Journal of Chemistry JO - Science Journal of Chemistry SP - 102 EP - 121 PB - Science Publishing Group SN - 2330-099X UR - https://doi.org/10.11648/j.sjc.20251304.12 AB - Over the last ten years, cancer therapies have struggled with drug resistance. In this report, we explore new coumarin (COU) compounds designed to inhibit the enzyme NQO1, which shows potential for effective treatment due to their favorable predicted drug properties. Three-dimensional (3D) models of NQO1-COUx complexes were generated through in situ modifications of the crystal structure of NQO1-COU12 (PDB entry code: 3JSX), which served as the reference compound for a training set of of 22 and a validation set of 6 VCOUs with known experimental inhibitory potencies. To identify the active conformation of COU1-22, we developed a gas-phase quantitative structure-activity relationship (QSAR) model that established a linear correlation between the calculated enthalpy of NQO1-COU complex formation and the values of experimental activities. Subsequently, we screened the Virtual Compound Library (VCL) using Lipinski's Rule of Five and the PH4 model, then assessed the potency of the new COU analogues using the retained QSAR model. The pharmacokinetic profile of the analogues obtained was also evaluated using the linear correlation equation derived from the QSAR model. The coefficient of determination (R²), the Leave One Out (LOO) cross-validated Squared and the Standard error of regression σ for this equation are 0.91, 0.94 and 0.14, respectively, thus revealing the high predictive power of this model. Similarly, the PH4 model, with a correlation coefficient of 0.91, demonstrated robust predictive power. A comprehensive screening of the COU virtual analogue library yielded a total of 63 drug candidates with oral bioavailability, among which the most promising compounds exhibited a predicted potency of 12.22 and a favorable pharmacokinetic profile. The integration of Quantitative Structure-Activity Relationship (QSAR) techniques and in silico screening, based on the PH4 model, has enabled us to propose potent anticancer candidates with optimal pharmacokinetic profiles. VL - 13 IS - 4 ER -
Laboratory of Fundamental and Applied Physics, University of Nangui Abrogoua, Abidjan, Côte d’Ivoire
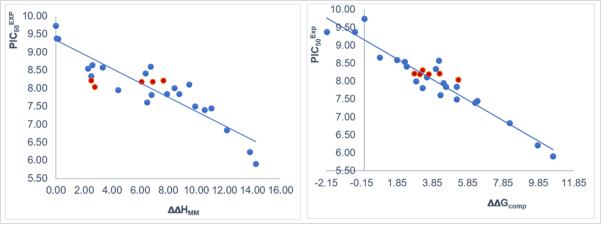
Figure 1. (Left) plot of correlation equation between and relative enthalpic contribution to the GFE (∆∆HMM [kcal.mol-1]). (Right) similar plot for relative complexation Gibbs free energies of the NQO1-COU complex formation ∆∆Gcom [kcal.mol-1] of the training set [9]. The validation set data points are shown in red color.
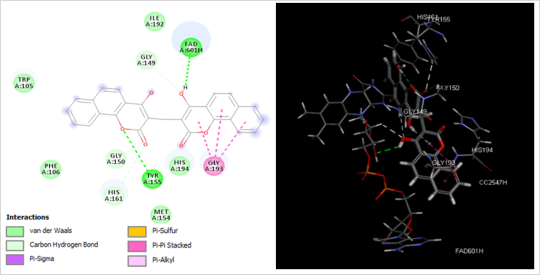
Figure 2. (Left) 2D schematic interaction diagram of the most potent inhibitor COU1 at the active site of NQO1 and (Right) 3D schematic interaction of COU1 at the enzyme active site.
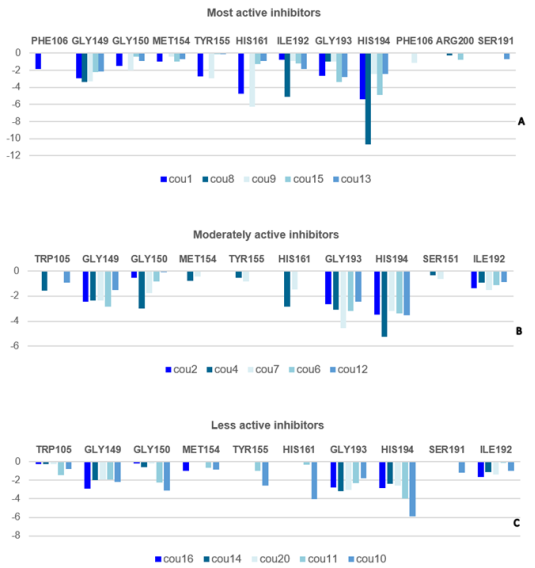
Figure 3. Molecular Mechanics intermolecular interaction energy Eint breakdown to residue contributions in [kcal.mol-1]: (A: Top) the most active inhibitors, (B: Middle) moderately active inhibitors, (C: Bottom) less active inhibitors, Table 2 [4].
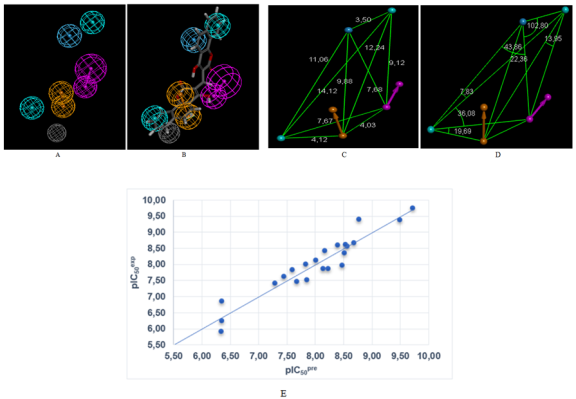
Figure 4. (A) Features coordinates of centers, (B) mapping of pharmacophore of NQO1 inhibitor with the most potent molecule COU1, (C) Distances between centers, (D) angles between centers of pharmacophoric features. Feature legend: HYD = Hydrophobic (cyan), HBA = Hydrogen bond Acceptor (green), HBD = Hydrogen bond Donor (pink), Excluded volume. (E) Correlation plot of experimental vs. predicted inhibitory activity.
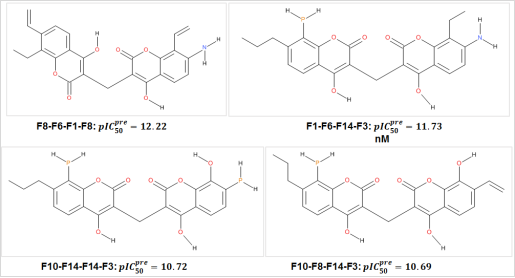
Figure 5. Best COU Analogs with scaffold of NQO1, the name is concatenation.
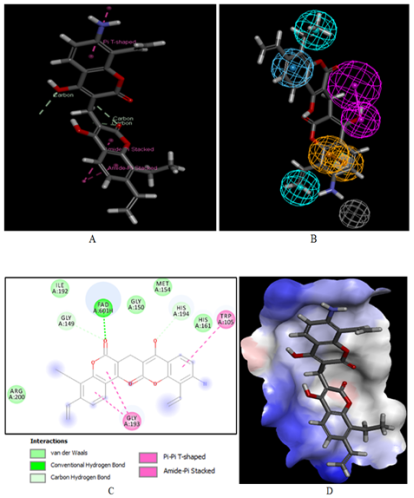
Figure 6. (A) Close up of virtual hit F8-F6-F1-F8, the most active designed COU analog () at the active site of NQO1. (B) Mapping of the COU F8-F6-F1-F8 to NQO1 inhibition pharmacophore. (C) 2D schematic interaction diagram of the best active designed COU analog F8-F6-F1-F8 at the active site of NQO1. (D) Surface of the active site of NQO1 with bound best active designed COU analog. The binding site surface is colored according to residue hydrophobicity: red = hydrophobic, blue = hydrophilic, and white = intermediate.
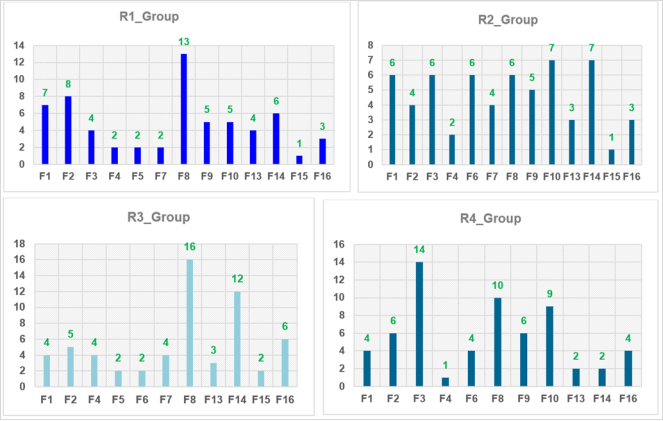
Figure 7. Histograms of frequency of occurrence of individual R-groups in the 63 best selected analogs mapping to four features of the PH4 pharmacophore hypothesis Hypo1.
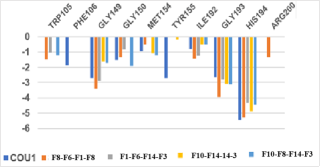
Figure 8. Molecular mechanics inter-molecular interaction energy Eint break-down to active site residue contributions in [kcal.mol-1]: the best four novel designed COU analogs (the color coding refers to ligands given in the legend).
Information




